Chief Researcher Officer
Synteny Biotechnology
Biography
I am the Chief Research Officer at Synteny Biotechnology. Our organisation uses modern AI methodologies combined with high-throughput experimental techniques to understand the specificities of T-cells, and in doing so, enable a new generation of T-cell based therapies and diagnostics. The company was founded by Lilly Wollman and Jamie Blundell, and I joined in January 2022 to build the organisation from scratch. By working at Synteny, I am able to focus on researching the fascinating natural computation that our bodies perform to identify pathogens and dysfunctional cells.
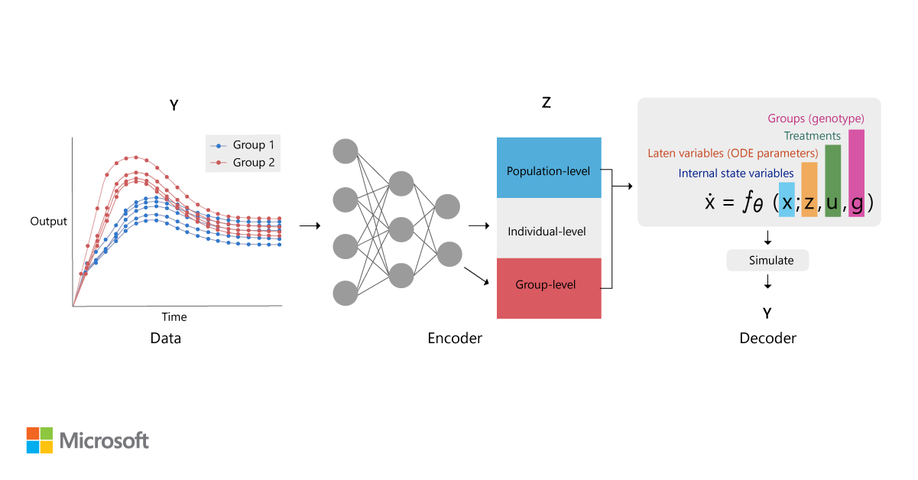
Before Synteny, I was a Principal Scientist and Research Manager at Microsoft Research Cambridge and project lead for Station B. Before that, I was a PhD student at University of Cambridge, where I worked on circadian timing in plants in the laboratory of Alex Webb.
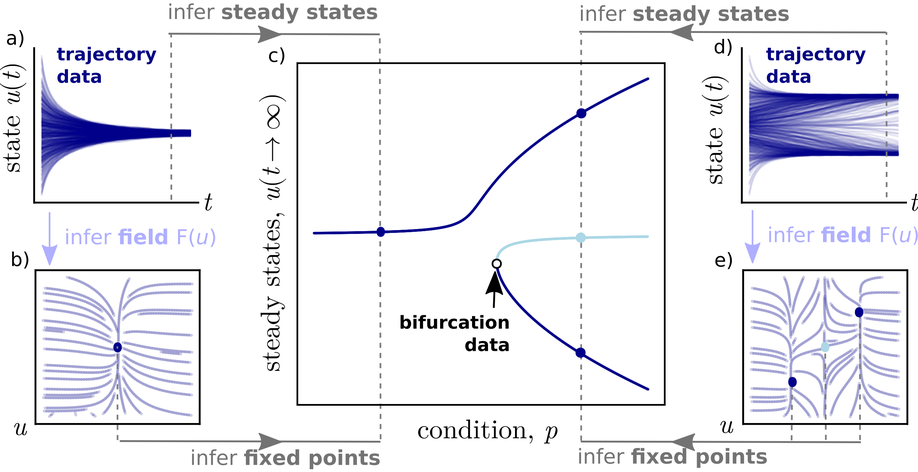
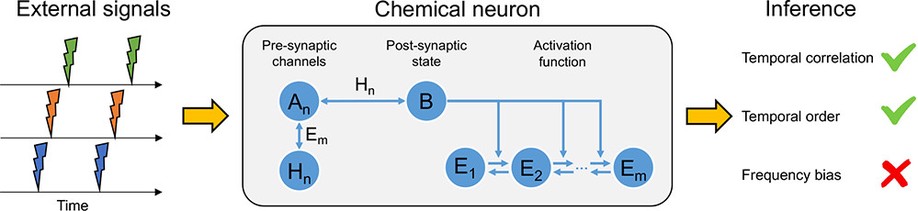
During my career, I have always operated at the intersection of biological data and computational analysis. I have made biological discoveries using a wide range of computational techniques. The majority of my earlier work used ordinary differential equation (ODE) models and stochastic chemical kinetics (essentially continuous-time Markov chains). I have also developed techniques for parameter inference and parameter synthesis with dynamical models. In my later years at Microsoft Research, I became interested in probabilistic machine learning models and active learning approaches, including Bayesian optimization. At Synteny, the primary observations are of amino acid sequences, and so I have become interested in using methods that can classify functional properties of those sequences, or repertoires (sets) of (T-cell receptor and antigen) sequences.
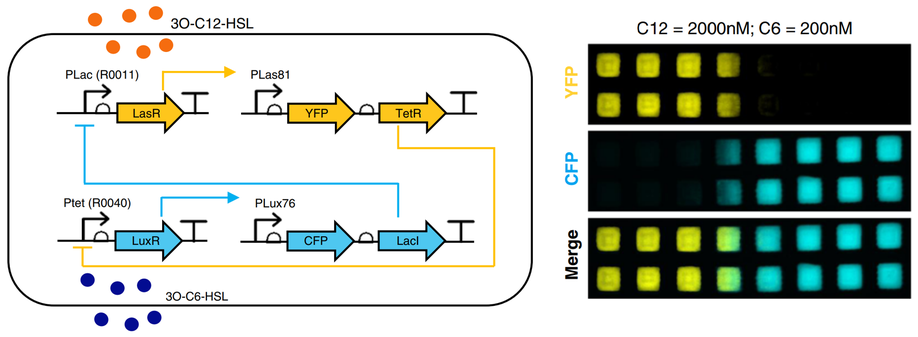
At Synteny, I have been able to reignite my interest in Immunology, which started when I first joined Andrew Phillips’ research group at Microsoft in 2009. Together, we developed some of the first dynamical models of antigen presentation by class I molecules of the major histocompability complex (MHC), collaborating with Tim Elliott, who at the time was at the University of Southampton.
Interests
- Immunology
- Synthetic Biology
- DNA Computing
- Bayesian Inference
- Bayesian Optimization
Education
-
PhD in Plant Sciences, 2009
University of Cambridge
-
MMath in Mathematics, 2005
University of Oxford